Analyse Single-cell RNA-Seq
De la sortie brute à un jeu de données entièrement annoté : contrôle qualité, suppression des doublets, intégration, clustering et annotation des types cellulaires, suivis des analyses aval qu'impose réellement votre biologie.
Échange confidentiel sous NDA · réponse sous 48 heures
Pourquoi c'est important
Le single-cell, ce n'est pas qu'une UMAP
Une UMAP n'est que le début de l'analyse. En mesurant l'expression gène par gène dans chaque cellule, le single-cell permet bien plus : isoler une population rare, comparer rigoureusement deux conditions, reconstruire une trajectoire de différenciation, relier un répertoire immun TCR/BCR à des états cellulaires. Mais ce signal ne sort pas seul des données : il faut les bonnes méthodes et de l'expertise biologique pour l'en extraire. C'est précisément notre métier, du contrôle qualité jusqu'aux analyses aval qui répondent à votre question. Vous repartez avec un résultat exploitable : une population à approfondir, une figure pour votre article, une réponse nette pour votre programme. Pas un dossier de fichiers de plus.
Pipeline analytique
Des reads bruts à des résultats exploitables
Chaque étape répond à des défis propres aux données single-cell : sparsité, ARN ambiant, doublets et effets de batch entre captures.
Quantification
FASTQ/BCL · Cell Ranger · split-pipe
QC & doublets
Empty drops · mito · scDblFinder
Intégration & clustering
Harmony · scVI · Leiden
Annotation
Marqueurs · référence · IA
Analyses aval
Pseudo-bulk DE · pseudotemps · TCR
Rapport & figures
Écrit · prêt à publier
Ce que nous proposons
Analyse single-cell, en profondeur
Le scRNA-seq est notre spécialité principale. Au-delà d'un workflow standard de clustering et d'annotation, nous menons régulièrement les projets vers les analyses qui répondent réellement à votre question biologique.
Annotation des types cellulaires
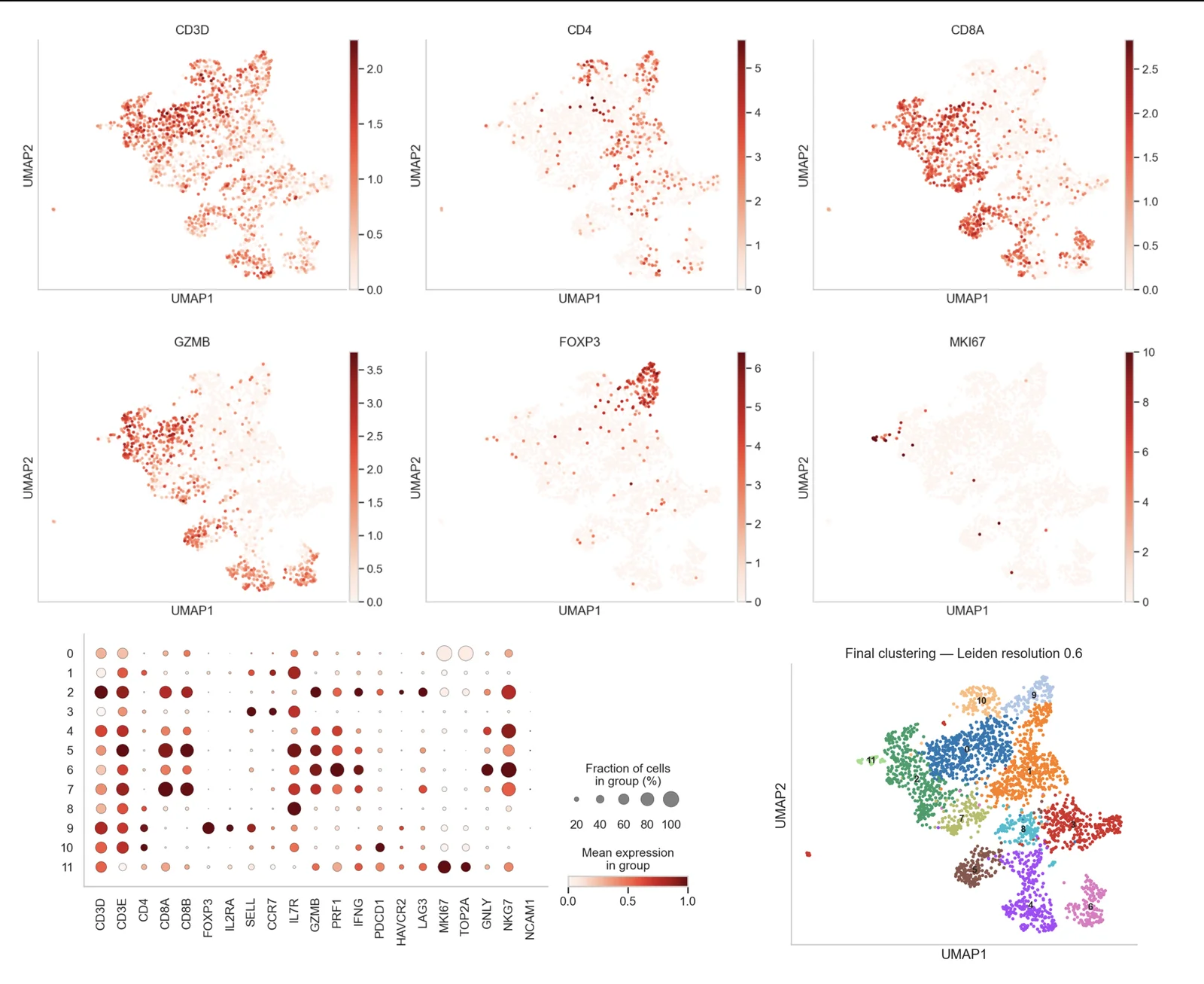
Annotation manuelle par marqueurs canoniques, par référence sur des atlas publics et assistée par IA, recoupée et validée par un expert.
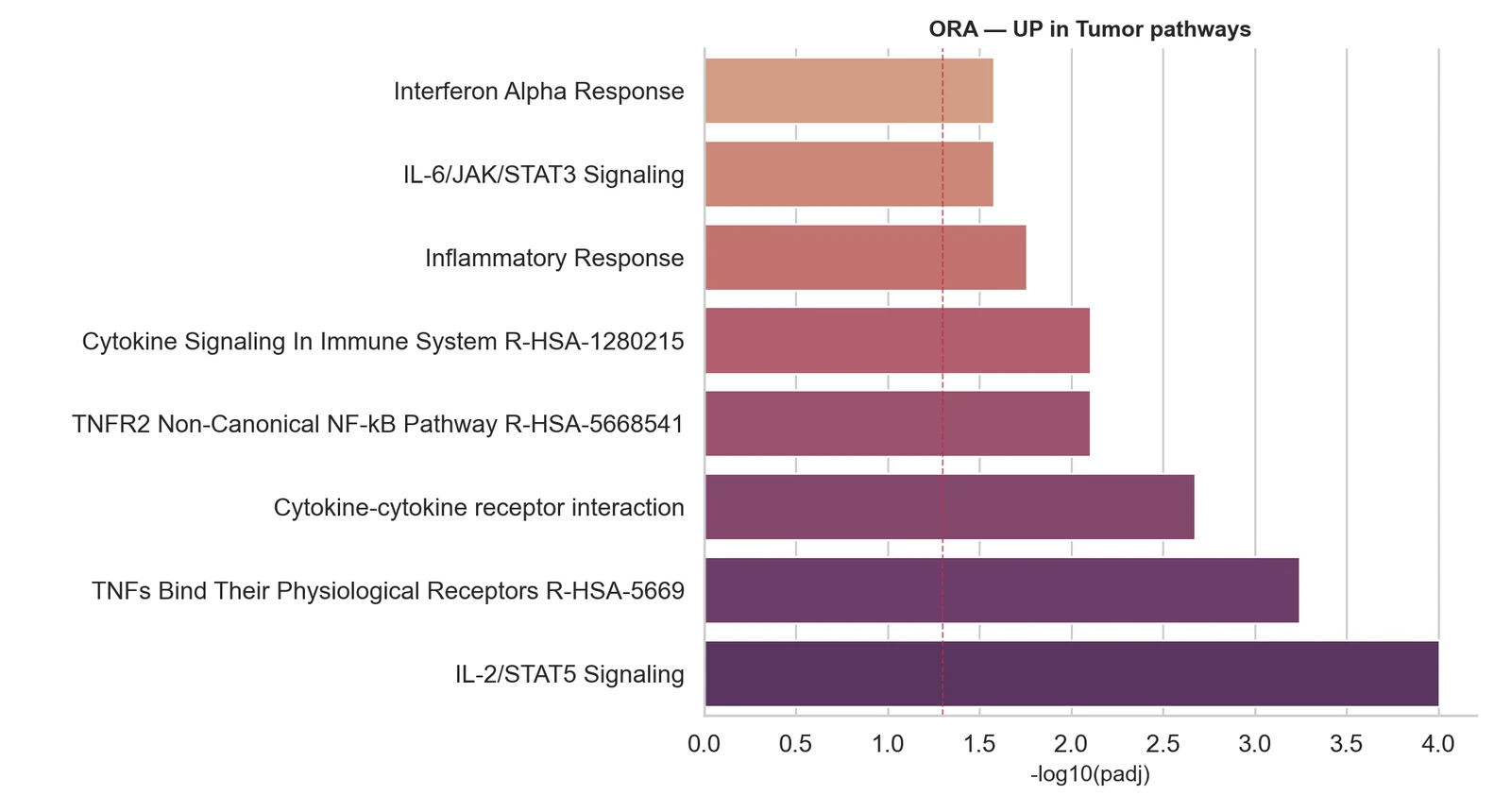
Expression différentielle pseudo-bulk
Comparaisons entre conditions : comptages agrégés par échantillon et type cellulaire, modélisés avec DESeq2 ou edgeR pour contrôler les faux positifs.
Trajectoires et pseudotemps
Trajectoires de différenciation et pseudotemps avec Slingshot, Monocle3 et PAGA, complétés par la RNA velocity (scVelo) pour la directionnalité.
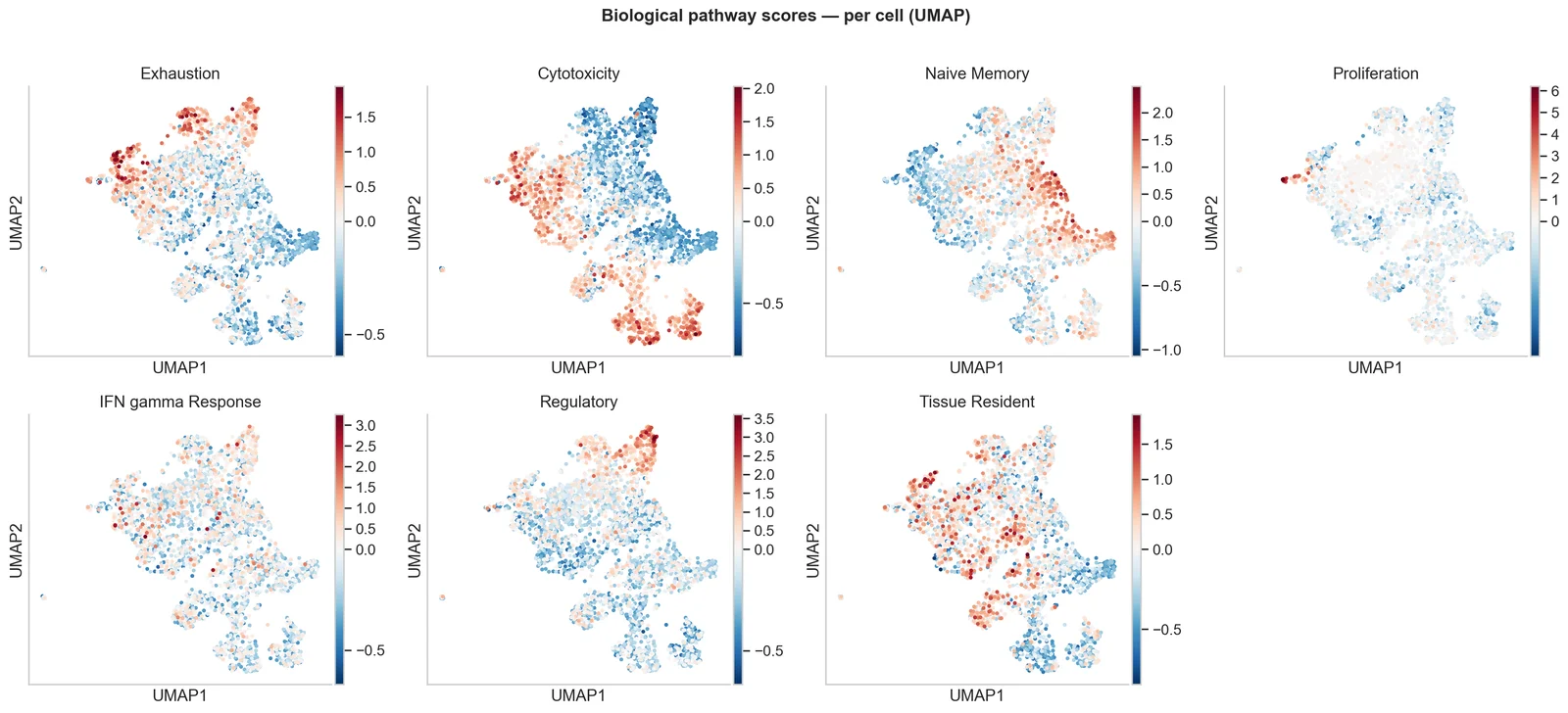
Inférence de voies et signatures
Activité des gene sets et des voies biologiques par cellule ou cluster, pour transformer des listes de marqueurs en programmes biologiques interprétables.
Profilage immun TCR/BCR
Expression génique single-cell et données VDJ appariées : clonotypes, expansion clonale et diversité du répertoire, reliés aux états transcriptionnels.
Intégration multi-omique
Protéines de surface CITE-seq, scATAC-seq (multiome), transcriptomique spatiale et RNA-seq bulk appairé combinés en une vision cohérente à travers les couches.
Le livrable
Des résultats structurés, interprétés et immédiatement exploitables
Les exemples ci-dessous illustrent des sorties typiques d'un projet single-cell. Le périmètre complet des livrables est toujours défini ensemble en amont : quelles analyses, quelles figures et quels formats servent le mieux vos objectifs scientifiques et votre budget.
Rapport écrit
Un rapport d'analyse complet couvrant les méthodes, les résultats et l'interprétation biologique, rédigé pour votre audience.
Session de restitution
Une présentation dédiée pour parcourir les résultats et répondre à vos questions.
Traçable
Versions des outils, paramètres et atlas de référence documentés pour chaque étape de l'analyse.
Contenu de l'analyse
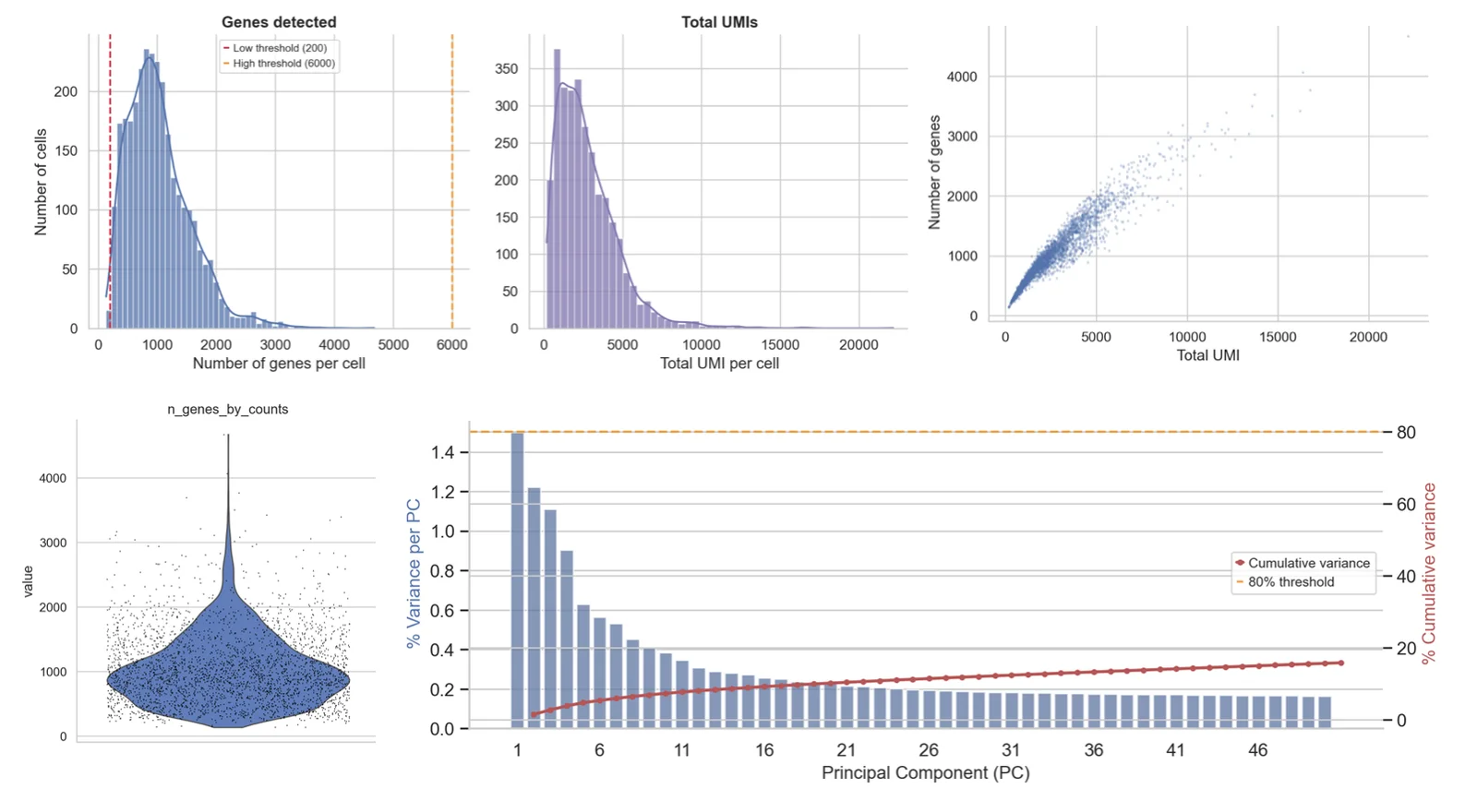
- QC & doubletsMétriques par cellule, filtrage, suppression des doublets.
- IntégrationCorrection de batch avec préservation du signal.
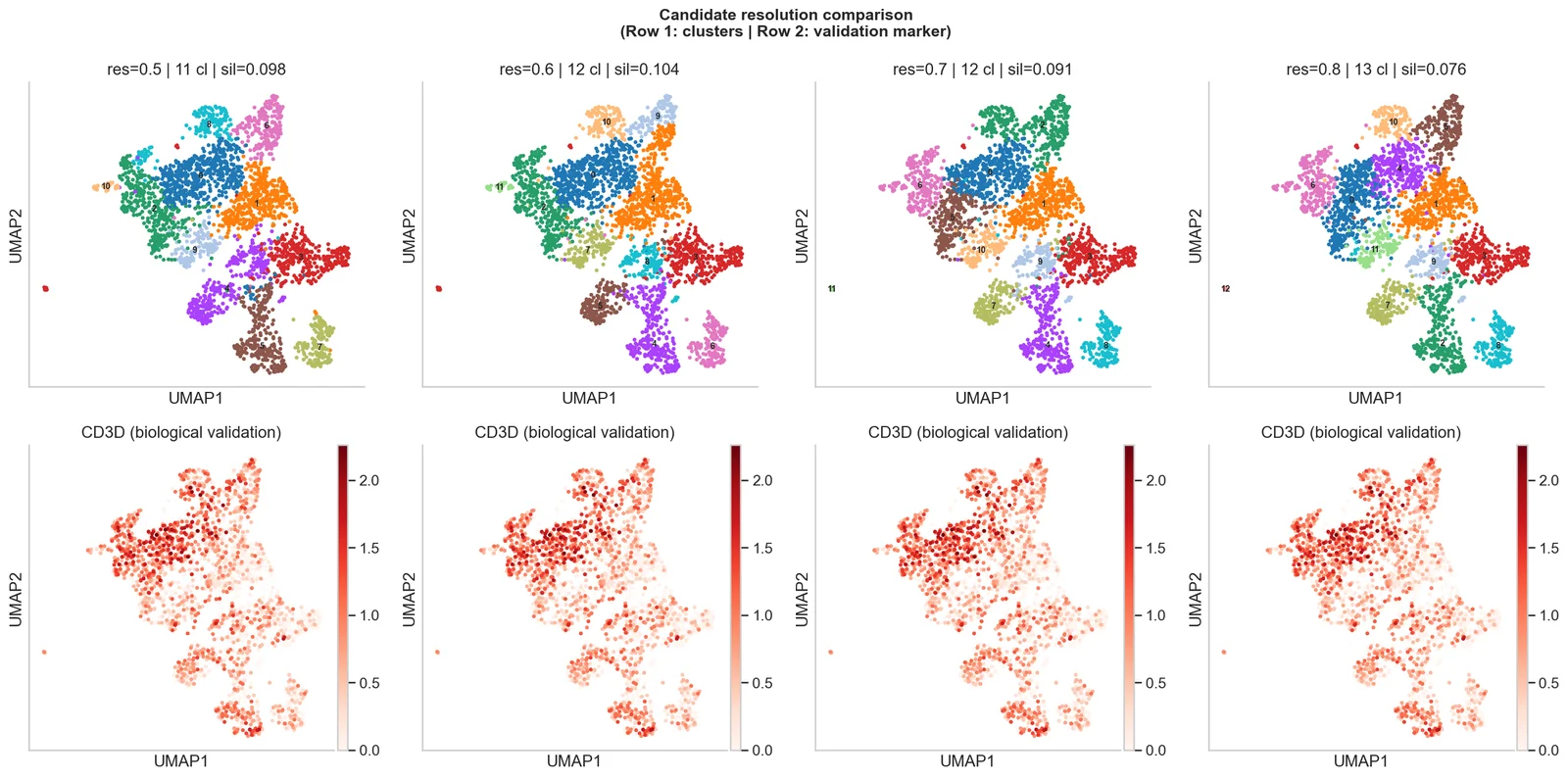
- Clustering & UMAPClusters Leiden et représentations.
- Annotation cellulaireÉtiquettes justifiées par les marqueurs.
- Gènes marqueursDotplots, heatmaps, tableaux par cluster.
- Expression différentielle (option)Pseudo-bulk par type cellulaire, DESeq2.
- Trajectoire / répertoire (option)Pseudotemps, RNA velocity, TCR/BCR.
- Méthodes & paramètresVersions des outils, références, reproductibilité complète.
Formats de livraison
- Rapport d'analyse : résultats, interprétation et conclusions.
- Figures prêtes à publier : haute résolution, formatées pour les journaux.
- Tableaux marqueurs & DE : symboles de gènes officiels, prêts à soumettre.
- Objet annoté :
.h5adou.rdspour votre propre réutilisation. - Session de restitution : présentation et questions/réponses en direct.
Notre méthode
Une rigueur analytique adaptée à la complexité du single-cell
Seuils de QC par cellule
Les seuils de fraction mitochondriale, de nombre de gènes et de profondeur UMI sont définis d'après la distribution réelle de vos cellules, pas des valeurs par défaut copiées-collées.
Intégration validée
La correction de batch est vérifiée sur des marqueurs biologiques de référence : nous confirmons qu'elle supprime le bruit technique sans écraser les vraies différences entre types cellulaires.
Annotation experte
Les labels automatiques et issus des référentiels sont systématiquement confrontés aux marqueurs par un expert qui connaît la biologie.
Pipeline versionné
Un workflow aux versions d'outils figées : la même entrée donne toujours le même résultat.
Les effets de batch en single-cell ne sont pas toujours corrigeables après le séquençage. Si votre étude est encore en phase de conception, notre analyse des situations où la correction computationnelle ne suffit plus est un bon point de départ, et notre conseil en bioinformatique couvre le design expérimental en amont de la génération des données.
Pour commencer
Plateformes et données que nous acceptons
Quel que soit votre point de départ (fichiers FASTQ bruts, matrice Cell Ranger ou objet partiellement annoté), le pipeline s'adapte pour reprendre là où vous en êtes.
Plateformes en gouttelettes
- 10x Genomics Chromium (3' et 5')
- Profilage immun 5' avec TCR/BCR VDJ
- BD Rhapsody et équivalents
Barcoding combinatoire
- Parse Biosciences Evercode (split-pipe)
- Smart-seq2 / Smart-seq3 en plaque
- Autres chimies personnalisées
Tout point de départ
- FASTQ ou BCL (sortie brute du séquenceur)
- Sortie Cell Ranger, 10x
.h5 - AnnData
.h5ad/ Seurat.rds
Questions fréquentes
Quelles plateformes single-cell prenez-vous en charge ?
Nous analysons couramment les données issues de 10x Genomics Chromium (3' et 5') et Parse Biosciences Evercode, ainsi que BD Rhapsody, Smart-seq2/3 et d'autres chimies de barcoding en gouttelettes ou combinatoire. Nous pouvons partir de la sortie brute du fabricant (Cell Ranger, split-pipe) ou d'une matrice de comptage déjà générée.
Partez-vous de fichiers FASTQ ou d'une matrice de comptage ?
Les deux. Nous pouvons traiter des fichiers FASTQ ou BCL bruts via les étapes de quantification (Cell Ranger, STARsolo, salmon alevin-fry, kallisto bustools ou split-pipe pour les données Parse), ou démarrer directement depuis une matrice ou un objet existant (10x .h5, .h5ad / AnnData, Seurat .rds) si la quantification a déjà été effectuée.
Comment gérez-vous les effets de batch et l'intégration multi-échantillons ?
Les effets de batch sont d'abord pris en compte au niveau du design expérimental, puis corrigés computationnellement avec des méthodes adaptées à votre structure (Harmony, scVI/scANVI, Seurat RPCA). Nous vérifions systématiquement que l'intégration supprime le bruit technique sans effacer le signal biologique, car certains confondeurs ne peuvent plus être corrigés après le séquençage. Nous détaillons cela dans notre article sur les effets de batch en scRNA-seq.
Comment annotez-vous les types cellulaires ?
Nous combinons trois approches complémentaires : annotation manuelle à partir des gènes marqueurs canoniques, annotation par référence sur des atlas publics (SingleR, Azimuth, CellTypist), et annotation assistée par IA pour accélérer et recouper les labels. Les étiquettes finales sont validées par un expert et justifiées par les marqueurs, jamais acceptées aveuglément depuis un appel automatique.
Pouvez-vous faire de l'expression différentielle entre conditions ?
Oui. Pour les comparaisons entre conditions, nous utilisons l'approche pseudo-bulk : les comptages sont agrégés par échantillon et type cellulaire, puis modélisés avec DESeq2 ou edgeR. Cette approche contrôle bien mieux les faux positifs que les tests naïfs cellule par cellule. Nous fournissons également les gènes marqueurs par cluster et, le cas échéant, une analyse de la composition en types cellulaires.
Proposez-vous des analyses de trajectoire, de pseudotemps et de RNA velocity ?
Oui. Nous inférons les trajectoires de différenciation et le pseudotemps avec des outils comme Slingshot, Monocle3 et PAGA, et pouvons ajouter la RNA velocity (scVelo) pour indiquer la directionnalité. Ces analyses sont appliquées quand elles répondent à une question biologique concrète, pas à titre de décoration.
Pouvez-vous analyser les données de répertoire immun TCR ou BCR ?
Oui. Nous traitons les données appariées d'expression génique single-cell et VDJ (10x 5' immune profiling et équivalents) : caractérisation des clonotypes TCR/BCR, expansion clonale, diversité du répertoire et mise en relation avec les états transcriptionnels. C'est un domaine de compétence particulier.
Pouvez-vous intégrer des données single-cell avec d'autres modalités ?
Oui. Nous intégrons le scRNA-seq avec les protéines de surface CITE-seq, le scATAC-seq (multiome), la transcriptomique spatiale et le RNA-seq bulk appairé, pour construire une image cohérente à travers les différentes couches omiques.
Comment la confidentialité de mes données est-elle garantie ?
Nous signons un accord de confidentialité avant tout transfert, échangeons les données via des canaux sécurisés et ne partageons ni ne réutilisons jamais vos données. Lorsque votre institution impose des exigences de sécurité spécifiques, nous nous y conformons pleinement.
Combien coûte une analyse scRNA-seq ?
Le tarif dépend de la plateforme, du nombre d'échantillons et de cellules, et de la profondeur des analyses aval souhaitées (annotation seule, ou avec expression différentielle, trajectoires, répertoire immun et intégration multi-omique). Chaque projet est cadré et devisé de façon transparente avant tout démarrage. Envoyez-nous votre design et votre type de données et nous vous répondons avec un périmètre précis, un calendrier et un devis forfaitaire sous 48 heures.
Prêt à transformer vos données single-cell en résultats ?
Envoyez-nous votre plateforme, votre design et votre type de données, nous répondons avec un périmètre clair, un calendrier et un devis forfaitaire transparent.
Échange confidentiel sous NDA. Réponse sous 48 heures.
Demander un devis