Analyse RNA-seq bulk
Quantification, expression différentielle et enrichissement de voies. Vos données analysées, interprétées et livrées sous forme de rapport interactif complet, pas une simple liste de gènes. De la biologie exploitable, que l'étape suivante soit une publication ou une décision R&D.
Échange confidentiel sous NDA · réponse sous 48 heures
Pourquoi c'est important
Un tableur de 20 000 gènes n'est pas un résultat
La plupart des projets RNA-seq calent au même endroit : une matrice de comptage brute et une liste de milliers de gènes que personne n'a le temps d'interpréter. Nous menons vos données de séquençage jusqu'au bout, avec des statistiques rigoureuses, les bons contrastes et des résultats replacés dans leur contexte biologique. L'objectif : aboutir à des conclusions, une figure pour votre article, une cible à prioriser, un go / no-go clair pour votre programme. Pas davantage de fichiers.
Pipeline analytique
Un workflow transparent, de bout en bout
Chaque étape repose sur des outils standards et validés par les pairs, avec des paramètres justifiés par vos données, jamais des valeurs par défaut arbitraires.
FASTQ
Reads bruts ou matrice de comptage
QC & Trimming
fastp · Q30 · stats par échantillon
Quantification
Alignement Salmon ou STAR
Expression différentielle
DESeq2 · modélisation robuste
Enrichissement
GSEA / ORA · Hallmarks, GO, KEGG
Rapport HTML
Interactif · autonome
Le livrable
Un rapport interactif, immédiatement actionnable
Chaque analyse est livrée avec un dashboard HTML autonome : aucune installation, aucune licence, compatible tous navigateurs. Vos résultats clés sont visibles en quelques secondes et prêts à partager avec votre équipe ou votre comité de pilotage.
Instantané
Résultats clés visibles en quelques secondes. Aucun logiciel à installer, rien à configurer : il suffit d'ouvrir le fichier.
Partageable
Un seul fichier HTML à transmettre à un collaborateur, un PI ou un comité de pilotage, tout est embarqué.
Traçable
Versions des outils, paramètres et références génome documentés, avec un manifeste SHA-256 pour chaque sortie.
Contenu du rapport
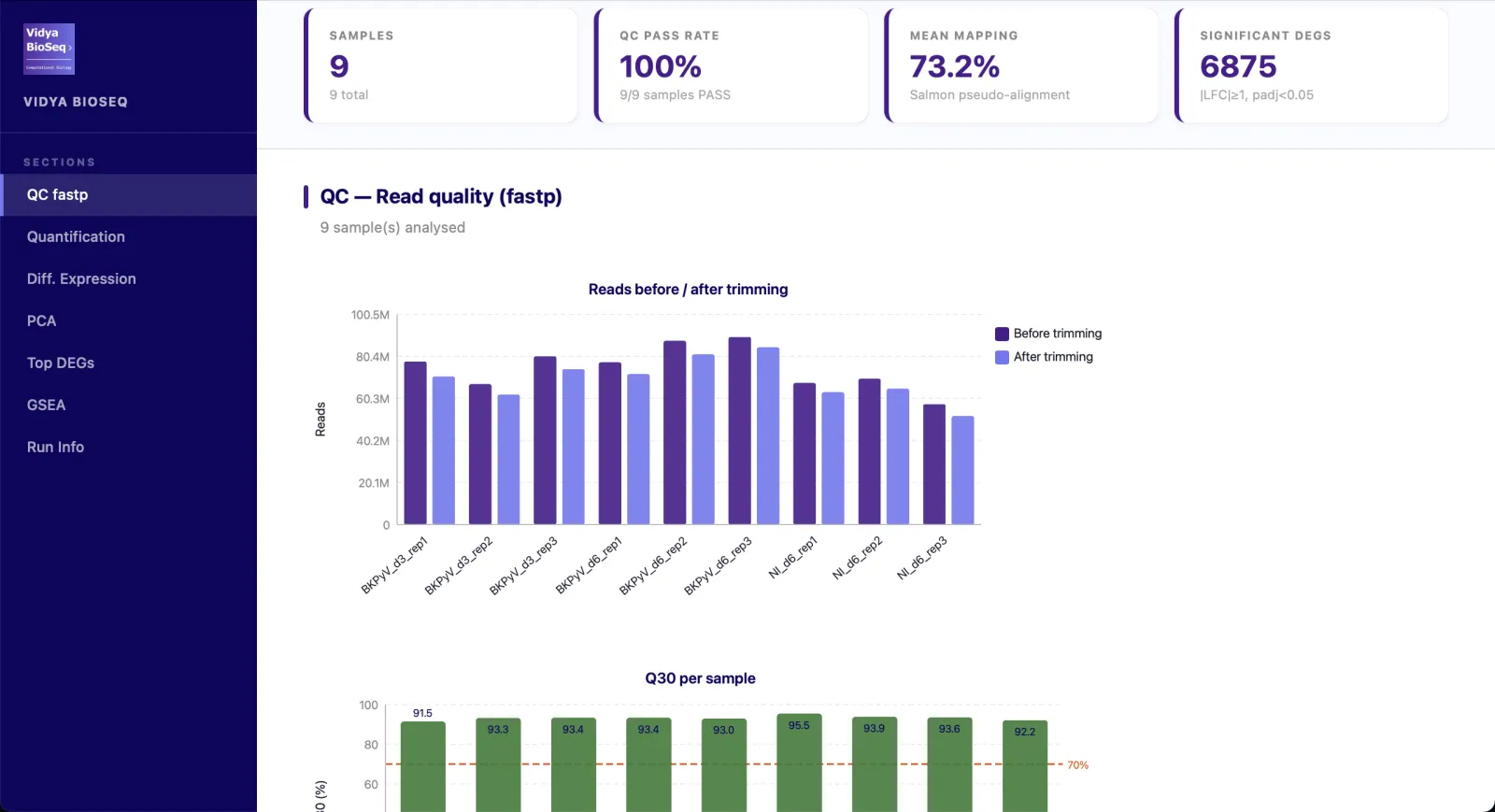
- QC & trimmingQualité des reads, Q30, stats fastp par échantillon.
- Quantification & alignementTaux de mapping par échantillon, Salmon ou STAR.
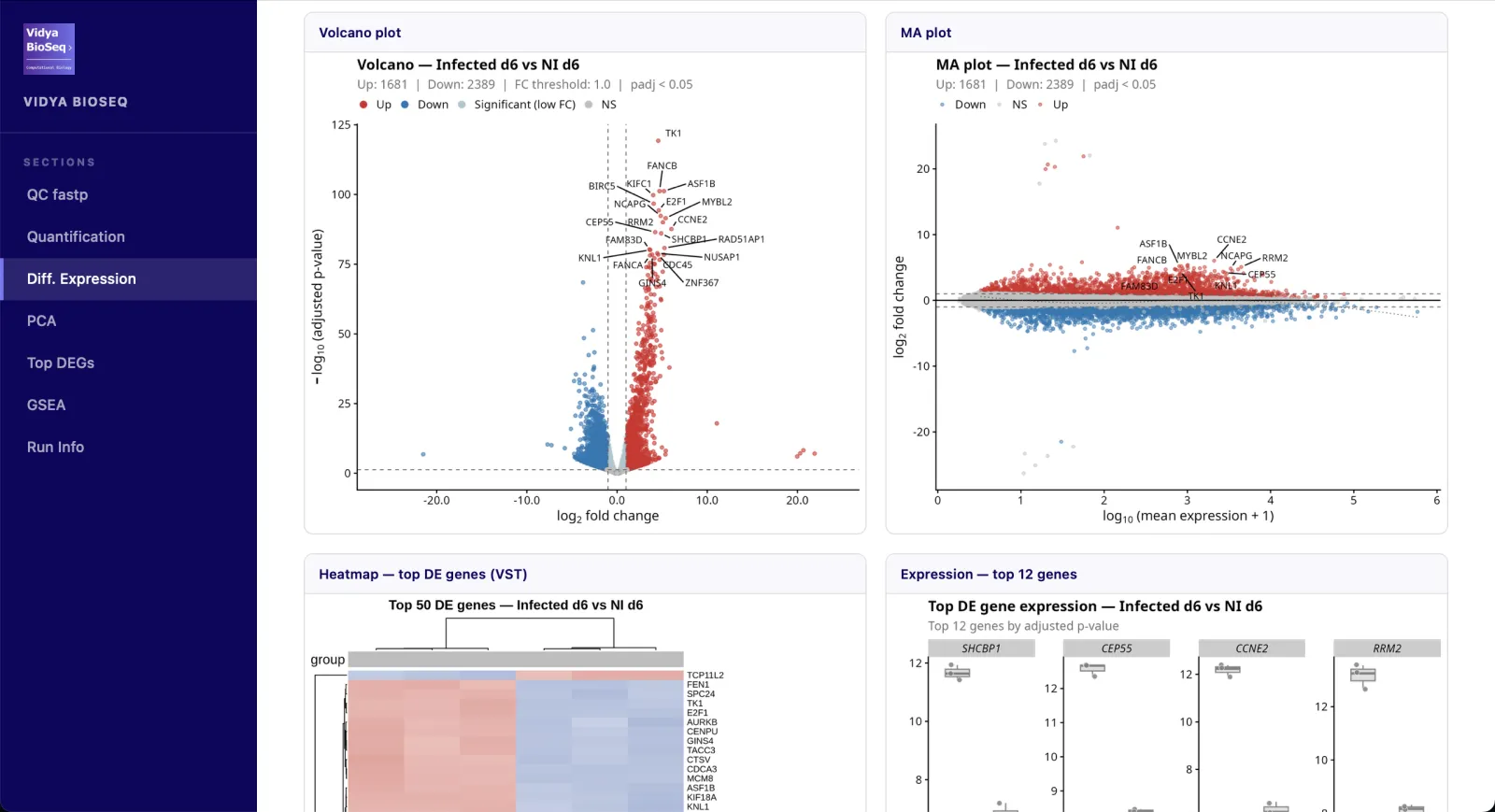
- Expression différentielleVolcano · MA plot · heatmap · stripplots.
- PCA & clusteringStructure des groupes et détection d'outliers.
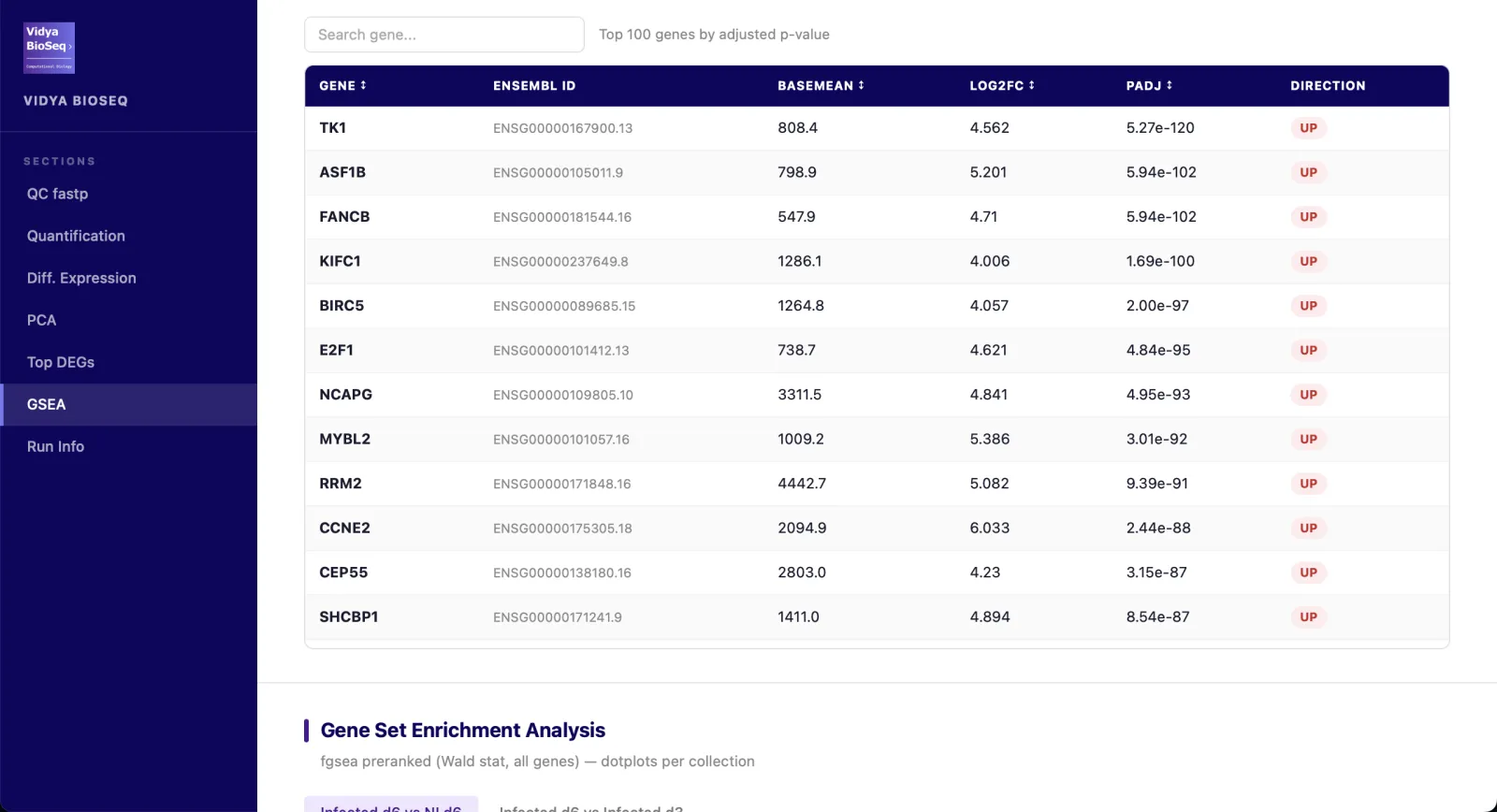
- Top DEGsTableau interactif trié par p-value ajustée.
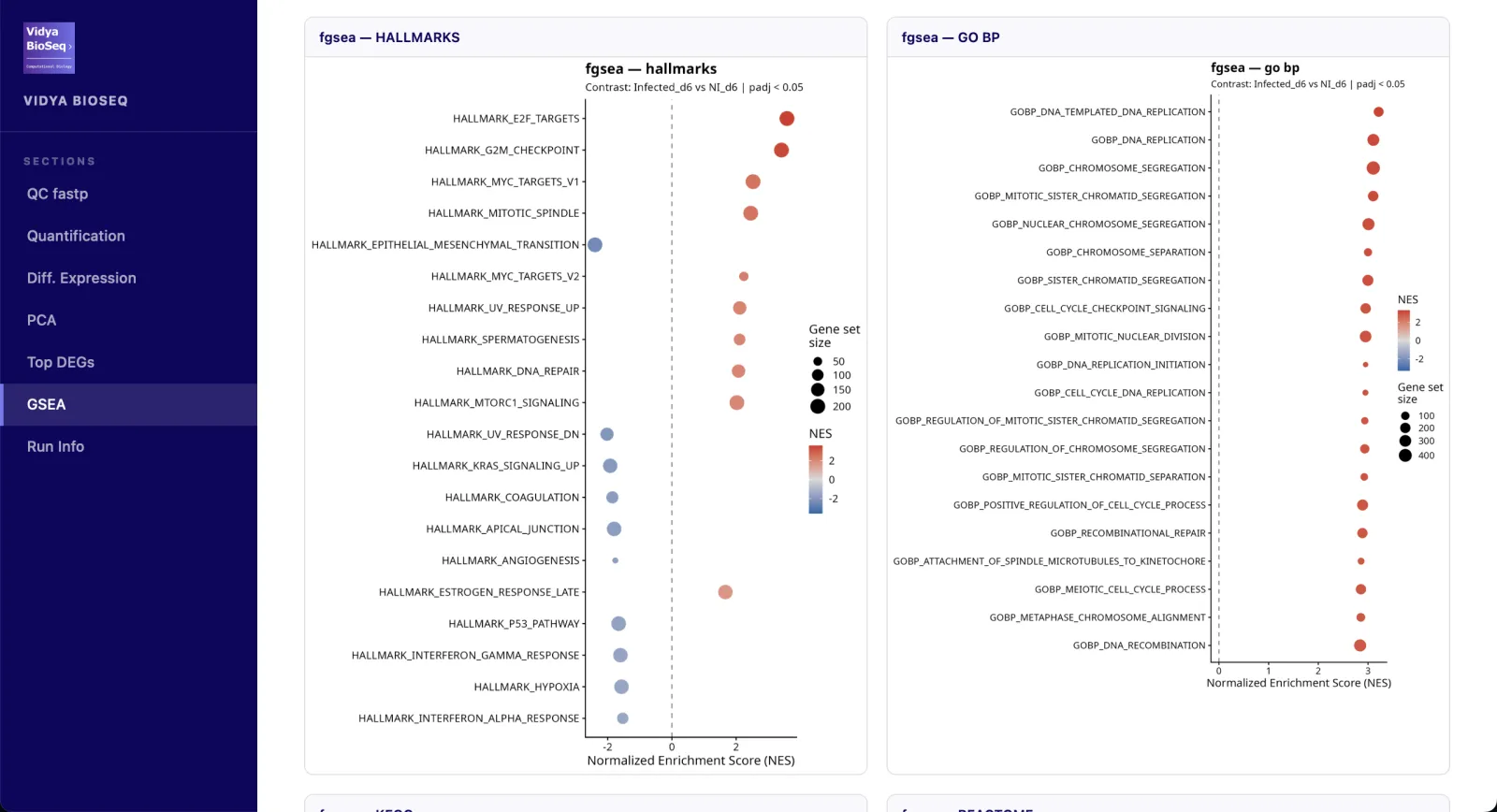
- GSEA / ORA (option)Hallmarks, GO-BP, KEGG, Reactome.
- Run infoVersions des outils, références, paramètres.
Formats de livraison
- Rapport HTML autonome : tout embarqué, s'ouvre hors ligne.
- Figures PDF haute résolution : prêtes à publier.
- Tableaux TSV annotés : symboles de gènes officiels (HGNC).
- Objet DESeq2
.rds: pour votre propre réutilisation. - Manifeste SHA-256 : intégrité de chaque fichier.
Notre méthode
Une rigueur défendable face à un reviewer ou à un comité R&D
Seuils pilotés par les données
Filtrage et seuils justifiés par la distribution réelle de vos données, pas des valeurs par défaut copiées-collées.
Pipeline versionné
Un workflow Snakemake aux versions d'outils figées : la même entrée donne toujours le même résultat.
Annotation lisible
Identifiants de gènes mappés vers les symboles officiels, pour des tableaux exploitables par des biologistes, pas seulement par des machines.
Interprétation biologique
Les résultats sont replacés dans leur contexte fonctionnel, transformant une liste de gènes en signature interprétable.
Pour commencer
Les données que nous acceptons
Quel que soit votre point de départ, nous reprenons votre projet, de la sortie brute du séquenceur à une matrice que vous avez déjà générée.
Reads de séquençage bruts
- Fichiers FASTQ, paired-end ou single-end
- Gzippés (
.fastq.gz) - Toute profondeur et longueur de reads
Matrice de comptage existante
- Matrices
counts.tsv/ CSV - Sortie featureCounts ou HTSeq
- Nous démarrons directement aux statistiques
Tout organisme
- Humain, souris, rat en standard
- Espèces non-modèles supportées
- Génome / annotation de référence personnalisés
Questions fréquentes
Partez-vous de fichiers FASTQ ou d'une matrice de comptage ?
Les deux. Nous pouvons traiter des fichiers FASTQ bruts (paired-end ou single-end, gzippés) via notre pipeline complet, ou partir d'une matrice de comptage existante (counts.tsv, featureCounts, HTSeq) si l'alignement a déjà été réalisé.
Combien d'échantillons et de réplicats faut-il ?
Au moins trois réplicats biologiques par condition sont recommandés pour une expression différentielle robuste. Moins de réplicats reste possible pour de l'exploratoire, et nous vous conseillons volontiers sur la puissance statistique avant de séquencer.
Quels organismes prenez-vous en charge ?
Toute espèce disposant d'un génome ou d'un transcriptome de référence : humain, souris et rat en standard, ainsi que les espèces non-modèles. Les identifiants de gènes sont mappés vers les symboles officiels (HGNC pour l'humain) pour des tableaux lisibles et prêts à publier.
Quel est le délai typique de réalisation ?
Un design standard (par exemple deux conditions avec réplicats) est généralement livré sous 72 heures après réception de vos données. Les designs plus complexes prennent plus de temps, et nous confirmons un calendrier précis avant de démarrer.
Pouvez-vous gérer des designs expérimentaux complexes ?
Oui. Les facteurs multiples, les effets de batch, les échantillons appariés et les cinétiques temporelles sont modélisés explicitement dans DESeq2, afin que les résultats d'expression différentielle reflètent la véritable structure de votre expérience plutôt qu'une simple comparaison à deux groupes.
Comment la confidentialité de mes données est-elle garantie ?
Nous signons un accord de confidentialité avant tout transfert, échangeons les données via des canaux sécurisés et ne partageons ni ne réutilisons jamais vos données. Lorsque votre institution impose des exigences de sécurité spécifiques, nous nous y conformons pleinement.
Et si j'ai besoin d'analyses au-delà du rapport standard ?
Le rapport standard est un point de départ. Nous ajoutons régulièrement des figures sur mesure, des contrastes supplémentaires, des signatures de gènes d'intérêt ou des analyses aval dédiées, et pouvons intégrer les résultats avec d'autres couches omiques si nécessaire.
Combien coûte une analyse RNA-seq bulk ?
Le tarif dépend de trois facteurs : le design expérimental (nombre de conditions et de contrastes), le nombre d'échantillons, et le niveau d'analyse souhaité. Une comparaison simple entre deux groupes avec un rapport interactif standard démarre généralement en dessous de 1 500 €. Les designs plus complexes, les bases de données de voies supplémentaires ou les analyses personnalisées sont chiffrés et devisés de façon transparente avant tout démarrage. Contactez-nous avec votre design et nous vous enverrons un devis forfaitaire sous 48 heures.
Prêt à transformer vos données RNA-seq en résultats ?
Envoyez-nous votre design et votre type de données, nous répondons avec un périmètre clair, un calendrier et un devis forfaitaire transparent.
Échange confidentiel sous NDA. Réponse sous 48 heures.
Demander un devis