Introduction
Chaque semaine, une équipe publie un résultat single-cell qui est en réalité un artefact de batch. La correction semble convaincante sur le UMAP. Les clusters sont propres et bien séparés. Le reviewer n'a rien vu. Les auteurs non plus.
Ce n'est pas un cas de figure rare. C'est l'un des angles morts de la transcriptomique single-cell, et l'un des moins discutés, précisément parce que le résultat a l'air correct.
Harmony, Seurat, ComBat, scVI… Les outils de correction de batch effects sont devenus une étape quasi automatique dans les pipelines d'analyse scRNA-seq. Ils sont puissants, bien benchmarkés, et dans de nombreuses situations, très efficaces. Mais ils partagent une limitation fondamentale qu'aucun talent technique ne peut surmonter : aucun algorithme ne peut sauver un design expérimental défaillant. Lorsque les sources de variation techniques et biologiques sont parfaitement confondues, la séparation n'est plus seulement bioinformatiquement difficile, elle devient mathématiquement impossible.
Comprendre où se situe cette frontière n'est pas un détail technique mineur. Cela détermine ce que vous pouvez, ou ne pouvez pas, conclure à partir de vos données.
La correction de l'effet batch est un outil computationnel puissant. Mais elle ne peut fonctionner que si la variabilité biologique et la variabilité technique ne sont pas parfaitement confondues.
Qu'est-ce qu'un effet batch en scRNA-seq ?
Un effet batch correspond à une variation systématique d'origine technique qui affecte les mesures d'expression génique entre groupes d'échantillons traités dans des conditions différentes.
Les sources les plus fréquentes incluent :
- Dissociation tissulaire — enzymes, durée d'incubation, température
- Capture cellulaire — plateforme, lot de réactifs, opérateur
- Préparation des librairies — lot d'enzymes de reverse transcription, cycles PCR
- Séquençage — flowcell, lane, profondeur de séquençage
Contrairement au bulk RNA-seq, le scRNA-seq présente deux difficultés supplémentaires :
- La composition en types cellulaires peut varier entre batches
- Le dropout stochastique introduit un bruit cellulaire important qui peut amplifier les différences techniques
L'existence d'un effet batch n'est pas problématique en soi. C'est un phénomène technique presque inévitable, et il existe des outils robustes pour le gérer. Le problème survient lorsqu'il devient inséparable du signal biologique d'intérêt.
Prenons un scénario réaliste : des biopsies tumorales, qui sont fragiles, sont dissociées 30 minutes plus rapidement que les tissus normaux appariés pour limiter la nécrose. Chaque cellule tumorale porte désormais une signature de stress différentielle par rapport aux contrôles. Après Harmony, le UMAP est propre. Mais des gènes de réponse au stress comme HSPA1A, JUN, FOS voient maintenant leurs expressions associées à ces tumeurs. Ils apparaîtront dans le tableau d'expression différentielle. Ils figureront dans l'article. Ils seront cités.
Ce n'est pas un cas hypothétique. Des variantes de ce scénario sont apparues à de nombreuses reprises dans la littérature. Et ce biais peut rester longtemps caché.
Panorama des méthodes de correction
Le champ des méthodes de correction de l'effet batch en scRNA-seq a considérablement mûri au cours de la dernière décennie. Ces méthodes opèrent à différents niveaux de l'analyse et reposent sur des hypothèses distinctes. Comprendre ces différences est essentiel à la fois pour choisir l'approche appropriée et pour reconnaître quand aucune ne suffira.
| Méthode | Approche | Niveau d'action | Forces & limites |
|---|---|---|---|
| Harmony | Soft clustering itératif dans l'espace PCA avec correction dépendante du batch | Très rapide et robuste. Résistant à la sur-correction. À considérer en première intention. Ne modifie pas la matrice de comptages. | |
| Seurat v4/v5 (CCA/RPCA) | Identification d'anchors entre datasets via CCA ou RPCA | Embedding / Matrice | Très utilisé. Peut mélanger des populations cellulaires si les compositions diffèrent fortement entre batches. |
| ComBat / ComBat-seq | Modèle bayésien empirique appliqué directement sur la matrice d'expression | Matrice corrigée | Bien caractérisé statistiquement. Performant dans plusieurs benchmarks. ComBat-seq préserve la nature en comptages. Moins adapté aux batch effects non-linéaires. |
| scVI | Modèle génératif profond (VAE) modélisant les comptages avec une distribution négative binomiale dans un espace latent | Modélise les comptages de manière probabiliste. Très scalable. Expression différentielle intégrée. Peut sur-corriger dans certains cas. | |
| scANVI | Extension semi-supervisée de scVI utilisant les annotations cellulaires | Meilleure conservation du signal biologique quand des labels fiables sont disponibles. Idéal pour le transfert de labels. | |
| MNN / FastMNN | Alignement basé sur les mutual nearest neighbors entre batches | Matrice corrigée | Principe élégant. Nécessite des populations cellulaires partagées entre batches. |
| BBKNN | Construction d'un graphe KNN équilibré entre batches | Graphe KNN | Léger et rapide. N'altère ni les comptages ni l'embedding, uniquement la structure de voisinage. |
Les benchmarks tels que scIB (Luecken et al., 2022, Nature Methods) montrent qu'aucune méthode n'est universellement optimale. Harmony et Seurat se distinguent pour les intégrations simples, tandis que scVI et scANVI sont préférés pour les intégrations complexes multi-études.
Le meilleur choix dépend de plusieurs facteurs : la nature de l'effet batch, la taille du dataset, le type de sortie requise (embedding vs. matrice corrigée) et la question biologique posée.
Les limites fondamentales de la correction
Toutes les méthodes de correction partagent une hypothèse clé : la variabilité biologique et la variabilité technique doivent être statistiquement séparables. Lorsque batch et biologie sont parfaitement corrélés, aucune correction fiable n'est possible.
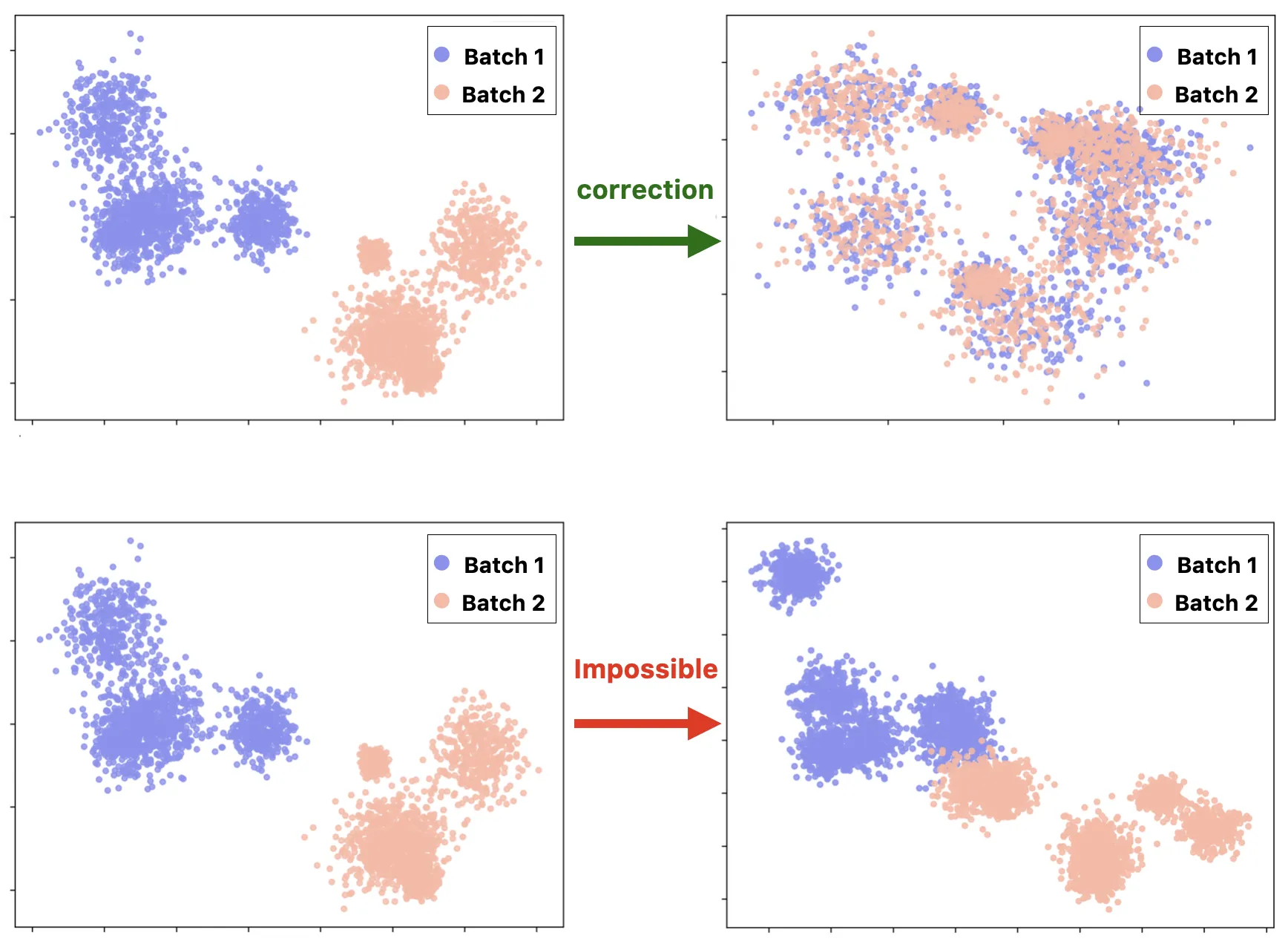
En pratique, nous rencontrons cette limitation fréquemment, et elle a généralement moins à voir avec le choix de l'algorithme qu'avec des décisions prises des mois plus tôt à la paillasse.
Le confounding : quand batch = biologie
Prenons un exemple simple :
- Cellules contrôle préparées le lundi (batch 1)
- Cellules traitées préparées le mardi (batch 2)
Dans ce cas :
- Ne pas corriger laisse un biais technique dans les résultats
- Corriger risque de supprimer le signal biologique d'intérêt
Les deux options sont décevantes. Il n'existe pas de troisième option qui fait disparaître le problème.
Ce constat a été formalisé par Song et al. (2020, Nature Communications), qui ont démontré que dans un design totalement confounded, aucune méthode ne peut séparer correctement les effets batch des effets biologiques. Ils ont également montré que certaines stratégies expérimentales, comme le reference panel design ou le chain-type design, permettent une correction valide précisément parce qu'elles brisent la corrélation parfaite entre batch et biologie.
L'intuition mathématique est directe : un modèle linéaire ne peut pas estimer simultanément deux variables parfaitement colinéaires. Ce que font les algorithmes dans ces situations n'est pas de la correction, c'est une partition arbitraire de la variance entre deux sources qu'on ne peut distinguer.
Si votre variable biologique d'intérêt (traitement, condition pathologique, etc.) est parfaitement confondue avec le batch, aucune méthode computationnelle, aussi puissante soit-elle, ne peut séparer les deux. Ce n'est pas une limitation logicielle ; c'est une impossibilité mathématique.
La sur-correction : quand la biologie disparaît
Le problème inverse est la sur-correction.
Des travaux récents ont montré que de nombreuses méthodes de correction peuvent altérer les données au-delà du nécessaire. Une correction excessive peut :
- Faire disparaître des gènes marqueurs connus
- Fusionner des populations cellulaires biologiquement distinctes
- Réduire artificiellement le nombre de gènes différentiellement exprimés
Certaines méthodes semblent plus résistantes à la sur-correction que d'autres. Harmony, par exemple, opère localement dans l'espace PCA et tend à préserver la structure globale. Les approches génératives profondes comme scVI sont plus expressives, ce qui est à la fois leur force et leur risque.
Le problème est que la sur-correction passe souvent inaperçue. Après correction, un UMAP qui semble biologiquement cohérent ne garantit nullement que la matrice d'expression sous-jacente reflète encore la biologie réelle. De nouveaux cadres d'évaluation comme RBET (Ye et al., 2025, Communications Biology) tentent de quantifier cela à l'aide de sets de gènes de référence, mais ces outils ne sont pas encore largement intégrés dans les pipelines standards.
Et les foundation models ?
L'émergence des foundation models single-cell (scGPT, Geneformer, Universal Cell Embeddings) a suscité l'espoir d'apprendre des représentations universelles capables de dépasser les batch effects. Le postulat est séduisant : entraîner des modèles sur des millions de cellules à travers des centaines d'études pour lui apprendra à encoder les types cellulaires dans un espace où la variation technique devient sans importance.
En pratique, le résultat reste encore imparfait. Lorsqu'on examine les représentations latentes de ces modèles, la structure de batch reste souvent visible, parfois atténuée, rarement éliminée. La raison n'est pas un défaut d'architecture. C'est un problème plus fondamental : les batch effects sont encodés dans les données d'entraînement elles-mêmes. Un modèle entraîné sur des données où « tumeur » et « réponse au stress élevée » co-occurrent ne peut pas apprendre à les dissocier, quel que soit son nombre de paramètres.
L'intégration d'objectifs de correction de batch directement dans l'entraînement des modèles est un domaine de recherche actif. Les résultats sont prometteurs mais préliminaires. Pour l'instant, les foundation models sont des outils puissants pour l'annotation cellulaire et la comparaison inter-études, mais ils n'éliminent pas le besoin d'un design expérimental rigoureux, et ils ne résolvent pas les confounded designs.
Le peer review ne détecte pas ce problème
Les reviewers voient typiquement le UMAP final, les annotations de clusters et les tableaux d'expression différentielle. Ils voient rarement la matrice de design expérimental (le tableau qui révélerait si une condition biologique n'apparaît que dans un seul batch). Les métriques d'intégration comme kBET ou LISI ne sont toujours pas rapportées systématiquement dans les publications. Et un dataset confounded bien corrigé peut produire des métriques parfaitement acceptables.
Des études avec des confounded designs peuvent passer le peer review, produire des visualisations propres et générer des résultats cités, tout en rapportant ce qui est un artefact technique. C'est un problème structurel dans la manière dont les résultats single-cell sont parfois évalués et rapportés.
Le design expérimental : première ligne de défense
Le meilleur moment pour gérer un batch effect, c'est avant qu'il n'existe.
Un design expérimental rigoureux est incomparablement plus efficace que n'importe quelle correction computationnelle appliquée a posteriori. Et de manière cruciale, les décisions clés se prennent avant le séquençage.
Bonnes pratiques au laboratoire
Plusieurs stratégies permettent de minimiser les batch effects lors de la préparation expérimentale :
- Traiter tous les échantillons dans des conditions expérimentales identiques (même opérateur, mêmes lots de réactifs, même équipement, même timing)
- Randomiser le placement des échantillons sur les cellules de capture
- Utiliser des approches de multiplexing pour pooler des échantillons biologiquement différents dans un même run technique
Les designs qui permettent la correction
Quand le multiplexing n'est pas possible, deux stratégies expérimentales ont été formellement démontrées comme permettant une correction valide.
Le reference panel design inclut un échantillon de référence commun dans chaque batch. Cet ancrage partagé permet l'estimation et la soustraction des effets batch car il fournit un signal biologique qui devrait être identique d'un batch à l'autre, toute différence est par définition technique.
Le chain-type design s'assure qu'au moins une population cellulaire ou un échantillon est partagé entre des batches consécutifs, créant une chaîne de ponts biologiques à travers l'expérience.
Les deux stratégies reposent sur le même principe fondamental : chaque batch doit partager au moins un composant biologique avec un autre batch afin que les algorithmes puissent distinguer la variation technique de la variation biologique.
Avant de démarrer votre expérience, construisez un simple tableau de contingence batches × conditions biologiques. Si une condition n'apparaît que dans un seul batch, c'est un signal d'alarme majeur. Corrigez le design maintenant, pas après le séquençage.
Diagnostiquer un effet batch irrécupérable
Comment déterminer si un effet batch est au-delà de ce que la correction peut résoudre ?
Avant toute correction
Commencez par examiner la PCA ou le UMAP coloré par batch. Si les cellules se regroupent principalement par batch plutôt que par catégories biologiques attendues, c'est un signal d'alerte initial, mais pas nécessairement irrécupérable.
Le diagnostic critique est le tableau de contingence batch versus condition biologique. Si une case de ce tableau est à zéro (une condition qui n'apparaît que dans un seul batch), vous avez au minimum un design partiellement confounded. Si chaque condition n'apparaît que dans un seul batch, le design est totalement confounded et la correction bioinformatique ne peut pas aider efficacement.
Après correction
Même lorsque la correction semble réussie, une validation minutieuse est nécessaire. Un UMAP propre après intégration n'est pas la preuve d'une correction réussie, c'est la preuve que l'algorithme a convergé. Ce n'est pas la même chose.
Vérifiez que les gènes marqueurs connus restent correctement exprimés dans les populations attendues. Comparez les analyses d'expression différentielle réalisées avec et sans correction. Une forte réduction du nombre de gènes DE après correction devrait éveiller les soupçons : êtes-vous en train de supprimer du bruit, ou du signal ?
Les métriques quantitatives peuvent aussi aider :
- kBET — k-nearest-neighbor Batch Effect Test
- LISI — Local Inverse Simpson's Index
- ASW — Average Silhouette Width
- ARI — Adjusted Rand Index
Utilisez ces métriques, mais interprétez-les avec précaution. Elles mesurent le mélange des batches, pas la préservation biologique.
Signaux d'alerte de sur-correction
Les signaux d'alarme typiques incluent :
- Des gènes ribosomaux ou mitochondriaux dominant les marqueurs de cluster
- Des populations biologiquement distinctes fusionnant dans l'embedding corrigé
- Des sous-populations rares attendues disparaissant après correction
- Une forte diminution du nombre de gènes différentiellement exprimés
- Des gènes marqueurs de contrôle positif connus perdant leur spécificité cellulaire
Checklist avant de lancer votre analyse
Au stade du design expérimental
- S'assurer de l'absence de confounding entre batch et variable biologique d'intérêt
- Inclure un échantillon de référence dans chaque batch si le multiplexing n'est pas possible
- Standardiser les protocoles (opérateur, réactifs, timing) et documenter les déviations
- Enregistrer soigneusement toutes les métadonnées techniques — lots de réactifs, durées de dissociation, opérateurs, cellules de capture
Au stade de l'analyse
- Visualiser les données avant correction (PCA/UMAP coloré par batch et par condition)
- Construire et examiner le tableau de contingence batch × condition
- Essayer Harmony comme méthode d'intégration rapide en première intention
- Évaluer les résultats à l'aide de métriques quantitatives (kBET, LISI, ARI)
- Valider les gènes marqueurs canoniques connus avant et après correction
- Si nécessaire, tester scVI ou Seurat en alternatives
- Quand la composition cellulaire diffère fortement entre batches, envisager scANVI si des annotations fiables sont disponibles
Quand la correction échoue
- Accepter le diagnostic — éviter de forcer des corrections qui effacent le signal pour produire un UMAP plus propre
- Reformuler les questions biologiques en fonction des limitations du dataset (ex. analyses intra-batch, analyses restreintes aux populations partagées)
- Si scientifiquement justifié, envisager de refaire l'expérience avec un design amélioré
- Documenter la limitation de manière transparente dans toute publication
Conclusion
Les méthodes modernes de correction de batch effects sont puissantes, mais elles reposent sur une hypothèse critique : la biologie et la variation technique doivent être distinguables. Quand le design expérimental est totalement confounded, aucune méthode bioinformatique ne peut récupérer l'information perdue. L'algorithme tournera quand même. Le UMAP sera toujours propre. Les résultats seront toujours publiables.
C'est précisément ce qui rend ce problème dangereux.
Si vous planifiez une expérience, le tableau de contingence est le pérequis à tout collecte de cellule. Si vous analysez des données existantes, un diagnostic rigoureux clarifiera au moins ce que le dataset peut, et ne peut pas, révéler de manière fiable. La réponse honnête à cette question a plus de valeur qu'une figure soignée construite sur un design irrécupérable.
Les outils de correction de l'effet batch sont puissants — mais ils ne peuvent pas sauver un design expérimental défaillant. Un UMAP propre n'est pas une garantie. Parfois, c'est exactement l'inverse.
Références
- Tran HTN et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biology 21, 12 (2020).
- Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods 16, 1289–1296 (2019).
- Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 (2019).
- Haghverdi L et al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbours. Nature Biotechnology 36, 421–427 (2018).
- Lopez R et al. Deep generative modeling for single-cell transcriptomics. Nature Methods 15, 1053–1058 (2018).
- Xu C et al. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Molecular Systems Biology 17, e9620 (2021).
- Luecken MD et al. Benchmarking atlas-level data integration in single-cell genomics. Nature Methods 19, 41–50 (2022).
- Song F et al. Flexible experimental designs for valid single-cell RNA-sequencing experiments allowing batch effects correction. Nature Communications 11, 3274 (2020).
- Antonsson SE & Melsted P. Batch correction methods used in single-cell RNA sequencing analyses are often poorly calibrated. Genome Research 35, 1832–1841 (2025).
- Ye Z et al. Reference-informed evaluation of batch correction for single-cell omics data with overcorrection awareness. Communications Biology 8, 504 (2025).
- Tung PY et al. Batch effects and the effective design of single-cell gene expression studies. Scientific Reports 7, 39921 (2017).
- Luecken MD & Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology 15, e8746 (2019).
- Andreatta M et al. Semi-supervised integration of single-cell transcriptomics data. Nature Communications 15, 872 (2024).
